El módulo Bio.SeqIO de Biopython proporciona una amplia gama de interfaces simples y uniformes para ingresar y generar los formatos de archivo deseados. Este formato de archivo solo puede manejar las secuencias como un objeto SeqRecord . Las strings en minúsculas se utilizan al especificar el formato de archivo. Los mismos formatos también son compatibles con el módulo Bio.AlignIO . La lista de los formatos de archivo se proporciona a continuación:
| Formato de archivo | Descripción |
|---|---|
| abi | Es un formato de seguimiento de secuenciación para Applied Biosystem, lee la secuencia capilar de Sanger, incluidas las puntuaciones de calidad PHRED para las llamadas base. Cada archivo contiene solo una secuencia, por lo que no tiene sentido indexar el archivo. |
| abi-trim | Igual que el formato abi pero tiene un recorte de calidad con el algoritmo de Mott. |
| as | Lee las secuencias superpuestas o contiguas del archivo de ensamblaje ACE . |
| cif-átomo | Se utiliza para averiguar la secuencia parcial de la proteína por estructura en función de las coordenadas atómicas. |
| cif-seqres | Determina la secuencia completa de la proteína mediante la lectura de un archivo de información cristalográfica macromolecular (también conocido como mmCIF ) según lo define el registro _pdbx_poly_seq_scheme . |
| emblema | Formato de archivo plano EMBL (Protein and DNA seq file format), utiliza Bio.GenBank internamente. |
| rápido | Un formato de archivo de secuencia genérico, cada registro comienza con una línea que comienza con el carácter > seguida de otras líneas de secuencia. |
| fasta-2line | Interpretación estricta del formato de archivo FASTA sin ajuste de línea (es decir, usando dos líneas por registro). |
| rapido | Una variante de FASTA con Sanger utilizada para almacenar valores de calidad de secuencia PHRED con un ASCII de desplazamiento 33. |
| fastq-sanger | Alias para FASTA que tiene consistencia con BioPerl y EMBOSS |
| fastq-solexa | Interpretación original de Solexa/Illumnia del formato FASTA que se utiliza para codificar puntajes de calidad de solexa con ASCII con compensación de 64. |
| fastq-illumina | Versión Solexa/Illumnia 1.3 a 1.7 del formato FASTA utilizado para codificar puntajes de calidad PHRED con ASCII de compensación 64. |
| gck | Formato local utilizado por Gene Construction Kit. |
| banco de gen | Formato de archivo plano GenBank o GenPept . |
| GB | Alias de genbank , que tiene consistencia con NCBI Entrez Utilities. |
| yo G | El formato de archivo de IntelliGenetics parece ser el mismo que el formato de alineación MASE . |
| imgt | Formato de variante EMBL de IMGT , donde las tablas de características se permiten intencionalmente para tipos de características más largos. |
| punta | El formato de archivo UCSC nib para nucleótidos utiliza 4 bits (1-nibble) para representar un nucleótido (2 nucleótidos requieren un byte). |
| nexo | Formato de alineación múltiple NEXUS , también llamado formato PAUP . |
| pdb-seqres | Lee el archivo PDB (Banco de datos de proteínas) y determina la secuencia de datos completa por encabezado. |
| pdb-átomo | Determina la secuencia parcial de proteínas por estructura según la sección de coordenadas atómicas del archivo. |
| Doctor | Estos archivos son la salida de PHRED , utilizados por PHRAP y CONSED para la entrada. |
| pir | Variante FASTA creada por NBRF (Fundación Nacional de Investigación Biomédica) para la base de datos de Recursos de Información de Proteínas (PIR), actualmente arte de UniProt . |
| seqxml | Formato de archivo XML simple. |
| sff | Archivos binarios en formato de diagrama de flujo estándar creados por Roche 454 y máquinas de secuenciación IonTorrent/IonProton. |
| sff-trim | El formato de diagrama de flujo estándar (SFF) aplica el recorte a los archivos listados. |
| Snapgene | Formato local utilizado por SnapGene . |
| suizo | Texto sin formato Swiss-Pro también llamado formato UniProt . |
| pestaña | Archivo de secuencia simple separado por tabulaciones de dos columnas, cada línea almacena el identificador y la secuencia del registro. |
| calidad | Variante FASTA que tiene valores de calidad PHRED de secuenciación de ADN. |
| uniprot-xml | Reemplazo del formato UniProt XML del formato de texto sin formato SwissProt . |
| xdna | Formato local de DNA Strider y SerialCloner , utilizado por Christian Marck. |
A continuación, la implementación explica cómo analizar dos de los formatos de archivo de secuencia más populares, es decir, FASTA y GenBank .
RÁPIDO:
Es el formato de archivo más básico para almacenar datos de secuencia. Originalmente , FASTA era un paquete de software creado durante la evolución temprana de la bioinformática para la alineación de secuencias de proteínas y ADN, utilizado principalmente para buscar similitudes. A continuación se muestra un ejemplo simple de análisis del formato de archivo FASTA :
Ejemplo:
Para obtener el archivo de entrada utilizado, haga clic aquí .
Python3
# Import libraries
from Bio.SeqIO import parse
# file path/location
file = open('is_orchid.fasta')
# Parsing the FASTA file
for record in parse(file, "fasta"):
print(record.id)
Producción:
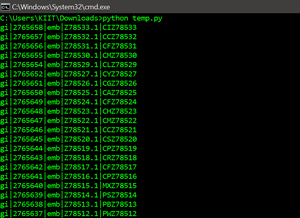
banco de genes:
Formato de secuencia más rico para genes que incluye varias anotaciones. Analizar el formato GenBank es tan simple como cambiar la opción de formato en el método de análisis de Biopython . A continuación se muestra un ejemplo simple de análisis del formato de archivo GenBank :
Ejemplo:
Para obtener el archivo de entrada utilizado, haga clic aquí .
Python3
# Import libraries
from Bio import SeqIO
from Bio.SeqIO import parse
# Parsing Sequence
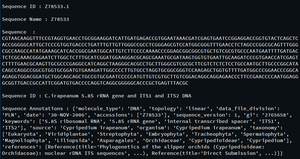
seq_record = next(parse(open('is_orchid.gbk'), 'genbank'))
# Sequence ID
print("\nSequence ID :", seq_record.id)
# Sequence Name
print("\nSequence Name :", seq_record.name)
# Sequence
print("\nSequence :", seq_record.seq)
# Sequence Description
print("\nSequence ID :", seq_record.description)
# Sequence Annotations
print("\nSequence Annotations :", seq_record.annotations)
Producción:
Publicación traducida automáticamente
Artículo escrito por jeeteshgavande30 y traducido por Barcelona Geeks. The original can be accessed here. Licence: CCBY-SA